Der Kauf einer Ausrüstung gibt Ihnen nicht die Erlaubnis, sie in einer pharmazeutischen Umgebung zu verwenden; Qualifizierung und Validierung stehen Ihnen noch im Weg. Ihr primäres Ziel ist es nicht, Sie von der Verwendung Ihrer Geräte abzuhalten oder deren Kosten zu erhöhen, sondern Ihnen zu helfen, eine gleichbleibende Qualität der Endprodukte zu gewährleisten.
Bei Sensum interagieren wir mit vielen pharmazeutischen Qualitätssicherungs-Teams zum Thema Qualifizierung und Validierung, da wir Lösungen für die automatische visuelle Inspektion von Endprodukten entwickeln und bereitstellen, die von der Qualitätssicherung stärker überwacht werden müssen als Qualitätskontrollsysteme. Seit mehr als 15 Jahren erleben wir unterschiedliche Qualifizierungsszenarien und konnten dabei mehrere bewährte Praktiken identifizieren. In den folgenden Abschnitten finden Sie praktische Einblicke in den Qualifizierungsprozess und einige nützliche Tipps, die Ihnen bei jedem Qualifizierungsprojekt helfen können.
Das wichtigste zuerst: Was ist Validierung und welche Qualifikation ist nötig?
Validierung ist ein umfassenderes Konzept als Qualifizierung und bezieht sich auf Vorgänge wie den Herstellungsprozess. Sie kann einfach als systematischer Ansatz erklärt werden, der Prozesse überprüft und ihnen hilft, erwartete und konsistente Ergebnisse zu erzielen. An der Validierung sind nicht nur Geräte beteiligt, sondern auch verschiedene ergänzende Systeme, Software und Personen, die Teil des Prozesses sind.
Die Validierung gliedert sich in mehrere Aktivitäten. Eine davon ist die Qualifizierung, die mit der Einführung von Systemen in den Prozess zusammenhängt. Die Aufgabe der Qualifizierung besteht darin, sicherzustellen, dass ein bestimmtes System den gesetzlichen Anforderungen, Industriestandards und der erwarteten Leistung entspricht.
Das (berüchtigte) V-Modell
Der Umfang der Qualifizierung richtet sich nach der Komplexität der Ausrüstung. Zum Beispiel sollte die Qualifizierung eines Schüttgutbehälters weniger Aufwand erfordern als die eines Sichtprüfsystems. Wir werden einen Blick auf die Qualifizierung eines konfigurierten computergestützten Systems werfen, das alle typischen Qualifizierungsschritte umfasst. Das Qualifizierungsverfahren für das Beispiel wird im folgenden V-Modell mit zwei Phasen, Spezifikation und Verifizierung, dargestellt.
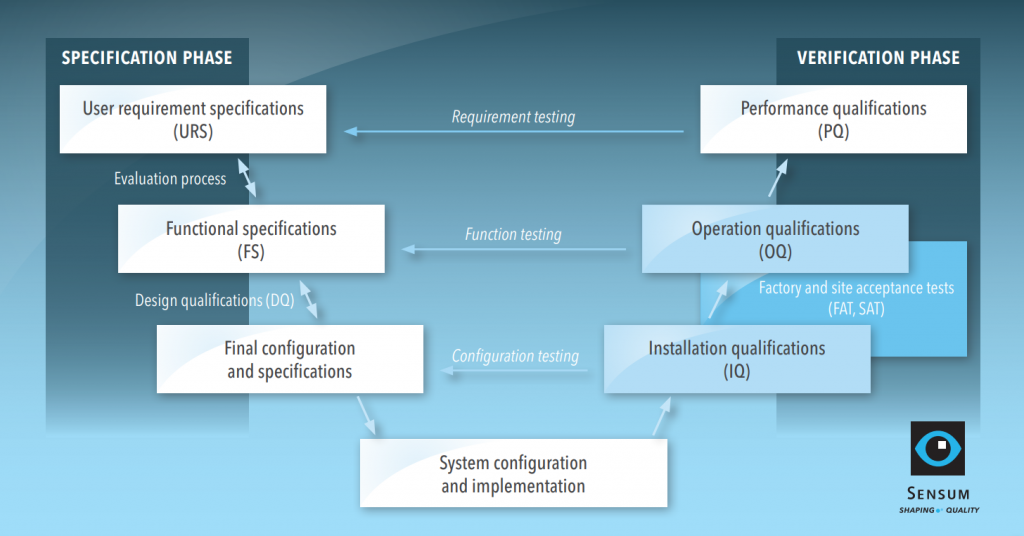
Spezifikationsphase: von URS bis DQ
Benutzeranforderungsspezifikationen (URS) werden vom Endbenutzer erstellt, der seine Erwartungen und Anforderungen an seinen Prozess auflistet. URS ist ein Basisdokument, das den gesamten Qualifizierungsprozess rationalisiert.
Bei Sensum als Lieferant stoßen wir auf viele URS. Die meisten URS-Dokumente enthalten viele Anforderungen mit mehr als 20 Seiten, aber die tatsächlichen Anforderungen, die für das spezifische Projekt relevant sind, werden in kaum ein oder zwei kurzen Punkten beschrieben. Dies liegt daran, dass die URS aus einer Vorlage oder aus der URS eines anderen Projekts ohne wesentliche Änderungen und Korrekturen erstellt werden. URS hat Auswirkungen auf den gesamten Qualifizierungsprozess und Abstriche sind hier nicht hilfreich. Einige TIPPS zu URS:
• Entfernen Sie allgemeine Anforderungen, die nicht anwendbar sind. Solche Anforderungen führen zu unnötigen Diskussionen oder erweitern sogar die Qualifikationen.
• Vergessen Sie nicht, die wichtigen Daten hinzuzufügen. Wenden Sie sich an Ihre Produktionsexperten, damit die Anforderungen die tatsächlichen Prozessanforderungen abdecken, wie z. B. Durchsätze, Betriebsqualität, Effizienz, Abfallerzeugung, Maschineneinstellungen, Teilemontage, Ausfallzeiten, Reinigung, Kalibrierung, Wartung, erforderliche Fähigkeiten zur Verwendung der Maschine usw.
Nachdem die URS vereinbart und genehmigt wurden, werden sie in der Regel mit mehreren potenziellen Lieferanten geteilt. Jeder Lieferant antwortet auf die URS mit einem Angebot und einer Reihe von Funktionsspezifikationen (FS), die schwer zu lesen sind und oft nicht mit den einzelnen URS-Punkten verknüpft werden können.
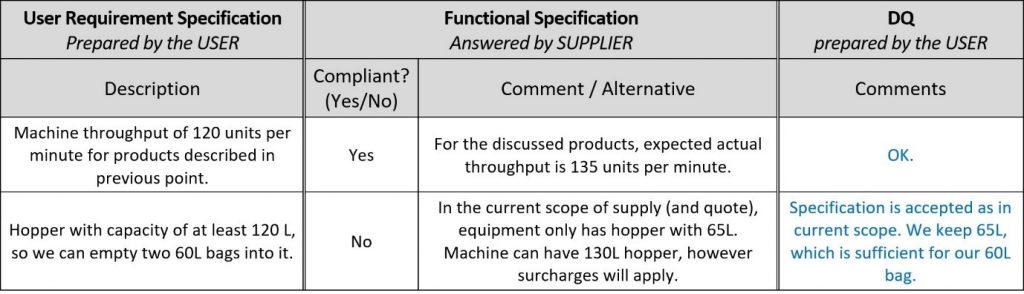
Um die Angebote der Lieferanten schneller bewerten zu können, sollten Sie im URS-Dokument Platz für ihre Kommentare schaffen und die neue Spalte „Funktionale Spezifikation“ nennen, denn ihre Kommentare sind in Wirklichkeit funktionale Bestätigungen und Beschreibungen ihrer Maschine! Auf diese Weise können Sie das vollständige Durchlesen der Konstruktionsunterlagen des Lieferanten vermeiden. Hier ist ein Beispiel:
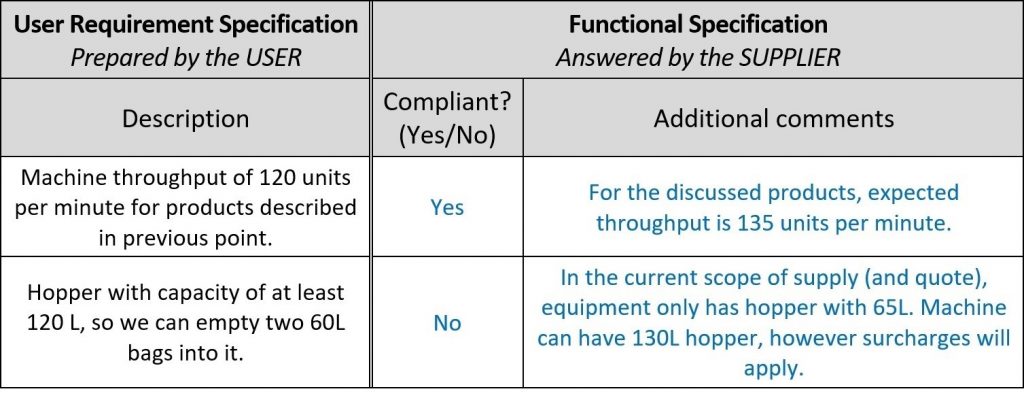
Sobald die Lieferanten ihr Feedback abgegeben haben, ist es Zeit für die Designqualifikation (DQ). Wie in der Einleitung erwähnt, hängt der Umfang der Qualifikationen von der Komplexität des Systems ab. In diesem Beispiel besteht die DQ aus drei Schritten: Angebotsbewertung, Risikoanalyse und Einrichtung von Tests. Was angesichts des großen Arbeitsaufwands problematisch klingt, bei ordnungsgemäßer Einrichtung jedoch beherrschbar ist.
Im ersten Schritt der DQ muss der Anwender prüfen, ob der Lieferant die in den URS beschriebenen Anforderungen erfüllt. Selbstverständlich gilt: Wenn ein Lieferant nicht alle Anforderungen erfüllen kann, sprechen Sie mit ihm und finden Sie für beide Seiten akzeptable Lösungen oder wählen Sie einen geeigneteren Lieferanten bzw. eine geeignetere Lösung. Wenn Sie die URS wie in diesem Artikel vorgeschlagen mit FS angehängt haben, kann ein Großteil der DQ durch Rückkommentieren der Kommentare des Lieferanten erledigt werden.
Führen Sie einfach eine DQ innerhalb des URS/FS-Dokuments durch, indem Sie Ihre Entscheidungen wie im folgenden Beispiel begründen.
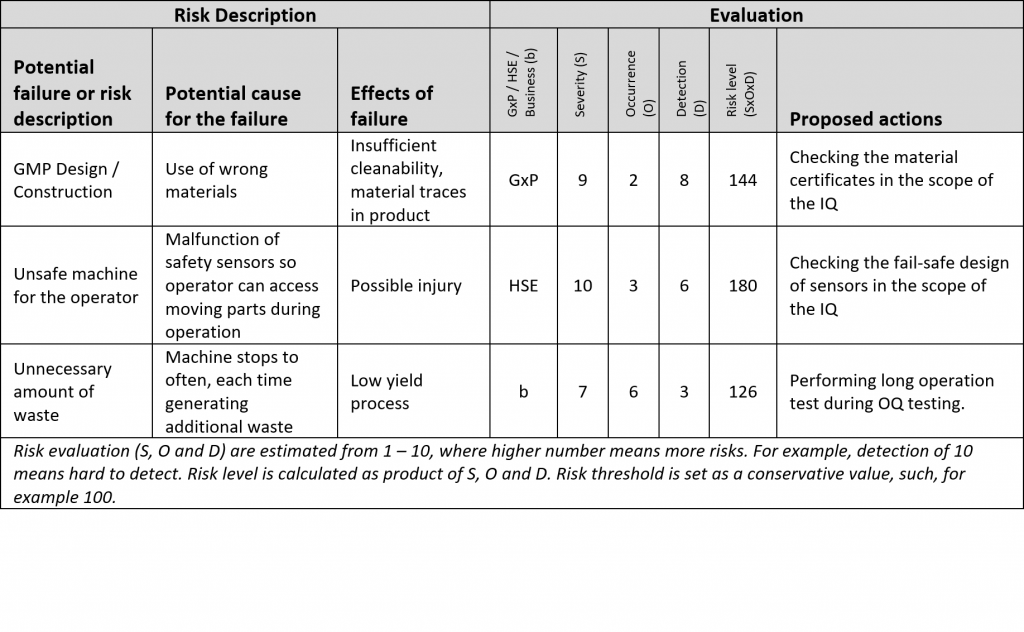
Der zweite Schritt der DQ ist die Risikoanalyse und wird erst gestartet, nachdem der erste Schritt zwischen Anwender und Lieferant vereinbart wurde. Das Ergebnis der Risikoanalyse sind Punkte und Spezifikationen, die bei der Qualifizierung geprüft und berücksichtigt werden müssen.
Die Risikoanalyse ist eine schwierige Aufgabe, insbesondere wenn die Technologie für den Benutzer neu ist. Versuchen Sie nicht, für jeden URS-Punkt ein mögliches Risiko zu fabrizieren. Nutzen Sie Erfahrung und gesunden Menschenverstand. Wenn Risiken aus irgendeinem Grund zu schwer zu definieren sind, sollte der Lieferant Ihnen bei der Risikoanalyse helfen können. Der Lieferant kennt die Lösung besser als jeder andere.
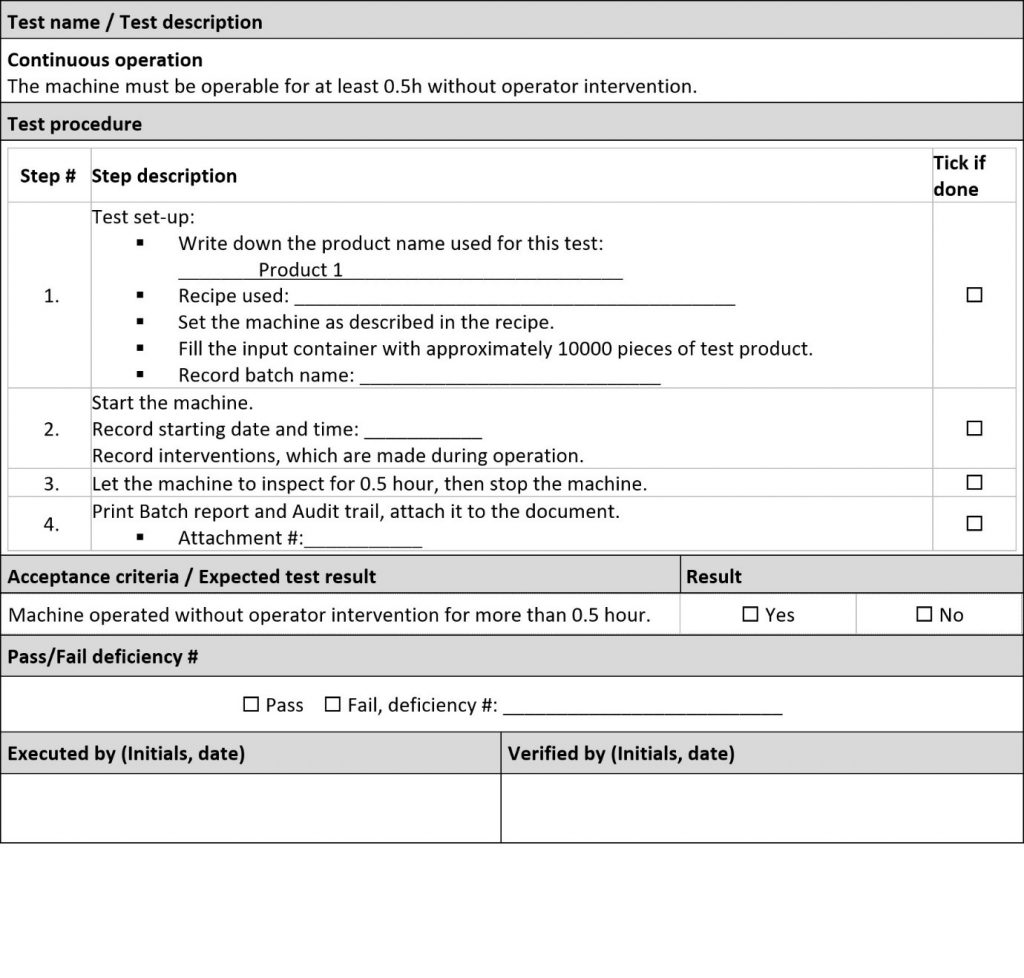
Der letzte Schritt der DQ ist die Einrichtung von Qualifikationstests für die Verifizierungsphase des V-Modells. Bei den Tests soll überprüft werden, ob der Lieferant wirklich alles wie vereinbart liefert, und alle Risiken angegangen werden, die über der Risikoschwelle liegen.
Das IQ/OQ-Dokument des Lieferanten umfasst Tests für die meisten erforderlichen Punkte und Risiken. Schauen Sie sich diese Tests zunächst an, bevor Sie mit der Einrichtung neuer Tests beginnen. Versuchen Sie außerdem, allgemeine Anforderungen und Risiken mit Funktionen zu begründen, um Ihre Qualifizierungsprotokolle zu vereinfachen und redundante Tests zu minimieren:
• Gehen wir von einem Risiko aus: „Eine Kamera im Inspektionssystem funktioniert nicht.“ Führen Sie keinen speziellen Test durch, um zu überprüfen, ob die Kamera installiert, an die Stromversorgung angeschlossen und funktionsfähig ist. Weisen Sie das Risiko einem allgemeinen Test zu, z. B. „Maschinenstart“, den Sie ohnehin durchführen werden, und begründen Sie, dass Sie nach dem Start Livebilder auf dem HMI sehen könnten und die Anlage daher über eine funktionsfähige Kamera verfügt.
•Nehmen wir nun eine Benutzeranforderung an den Prüfpfad an: „Alle Aktionen an der Maschine müssen im Audit-Trail aufgezeichnet werden.“ Führen Sie keinen speziellen Test „Audit-Trail prüfen“ durch. Versuchen Sie, die Anforderung einem Betriebstest zuzuordnen, bei dem der Batch-Bericht mit Audit-Trail aus anderen Gründen überprüft wird.
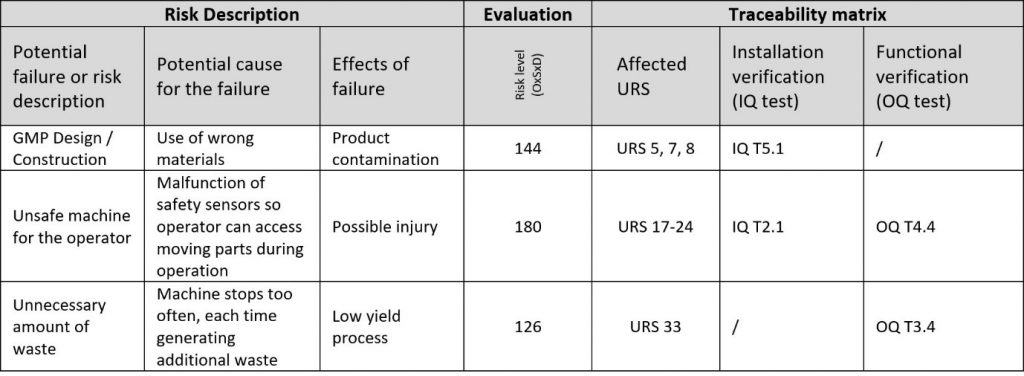
Das Endergebnis der DQ ist die Rückverfolgbarkeitsmatrix, die Risiken und Anforderungen mit Tests verknüpft.
Rückverfolgbarkeitsmatrizen sind für viele Dinge bekannt. Dem Projektteam Zeit zu sparen, gehört nicht dazu. Die Herausforderung besteht darin, Zusammenhänge zwischen URS, Risiken und Tests klar und so einfach wie möglich zu gestalten. Erfahrungsgemäß wird es immer mehr URS-Punkte als Risiken geben. Ordnen Sie daher URS-Punkte den Risiken zu und nicht umgekehrt. Einige URS-Punkte bleiben möglicherweise sogar nicht zugewiesen, was nur darauf hinweist, dass nicht zugewiesene URS-Punkte kein Risiko für das Projekt darstellen.
Verifizierungsphase: Zum Schluss IQ, OQ und PQ
Wenn die Spezifikationsphase abgeschlossen ist und der Lieferant für die Installation bereit ist, beginnt die Verifizierungsphase. Der Benutzer und der Lieferant befolgen die IQ/OQ-Protokolle und der Benutzer schließt die Qualifizierung mit PQ ab.
Benutzer und Lieferant sollten sich während der DQ auf das genaue Protokoll und den Umfang der Tests einigen, um die für beide Parteien riskante Erstellung neuer Tests während der Qualifizierung zu minimieren.
IQ/OQ wird normalerweise zweimal durchgeführt. Erstens erfolgt dies beim Lieferanten im Rahmen von Factory Acceptance Tests (FAT). Während der FAT sind Änderungen am System aufgrund von Anforderungsänderungen (die häufig vorkommen) oder aufgrund möglicher Abweichungen nicht so teuer wie später, wenn sich das System außerhalb der Produktionsanlagen befindet.
Bei den FAT handelt es sich in der Regel um die erste Erfahrung des Benutzers mit der Maschine. Verbringen Sie so viel Zeit wie möglich mit OQ, denn OQ besteht aus Tests, bei denen die Maschine ihre Aufgabe erfüllt. Es ist schwer vorstellbar, dass eine schlimmere Abweichung eine Sicherheits- oder Funktionsabweichung darstellt. Die IQ ist jedoch nach wie vor Voraussetzung für die OQ. Versuchen Sie also, sie so schnell wie möglich zu erreichen, indem Sie nur das Nötigste tun und weitere administrative Tests mit „N/A bei FAT“ oder „Nicht risikobehaftet, wird bei SAT geprüft“ auslassen, um so schnell wie möglich zur OQ zu gelangen.
Zudem wird IQ/OQ mit denselben Produkten nach der endgültigen Installation beim Benutzer im Rahmen von Site Acceptance Tests (SAT) wiederholt.
Senden Sie den Endbenutzer (Bediener aus der Produktion) an FAT/SAT. Wenn die Bediener die Tests durchführen, werden FAT und SAT gleichzeitig zu einem sehr effizienten Training. Versuchen Sie außerdem, die operativen Tests der OQ so zu gestalten, dass sie sich wie echte Abläufe/Prozesse anfühlen. Typische Kapitel im OQ sind:
• Hardware- und Software-Setup vor dem Start einer Charge
• Tests für Zuführung/Entladung
• Tests für Langzeitbetrieb, Leistungsbewertung, Abfallerzeugungsbewertung, Durchsatzkontrollen
• Ausfallzeittests: Reinigungstests, Montage-/Demontagetests
OQ-Erfahrungen sind bei der Vorbereitung auf PQ sehr willkommen.
Die Leistungsqualifizierung (PQ) wird vom Benutzer nach erfolgreichem SAT durchgeführt. Der Benutzer sollte eine Standardarbeitsanweisung (SOP) vorbereitet haben und diese während der PQ befolgen. Nach erfolgreicher PQ hergestellte/verarbeitete Produkte können bereits kommerziell genutzt werden.
Der Lieferant kann Ihnen bei der Optimierung Ihrer SOP helfen, die viele Jahre lang verwendet wird. Eine Optimierung und Änderung zu diesem frühen Zeitpunkt wird die Erfolgsquote der PQ und die Erfolgsquote aller späteren Läufe verbessern.
Anhänge
• Beispiel – IQ/OQ für Werksabnahmetests
• Beispiel – Vorlage für eine Risikobewertungs- und Rückverfolgbarkeitsmatrix
• Beispiel – Spezifikationen der Benutzeranforderungen – URS
Dieser Artikel wurde ursprünglich in Pharmaceutical Technology veröffentlicht:
– https://www.pharmaceutical-technology.com/sponsored/pharmaceutical-qualification-and-validation/
Similar posts